Scalable Prediction of Acute MyeloidLeukemia Using High-DimensionalMachine Learning and Blood Transcriptomics
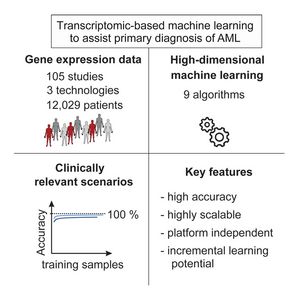
Acute myeloid leukemia (AML) is a severe, mostly fatal hematopoietic malignancy. We were inter-ested in whether transcriptomic-based machine learning could predict AML status without requiringexpert input. Using 12,029 samples from 105 different studies, we present a large-scale study of ma-chine learning-based prediction of AML in which we address key questions relating to the combinationof machine learning and transcriptomics and their practical use. We find data-driven, high-dimensionalapproaches—in which multivariate signatures are learned directly from genome-wide data with noprior knowledge—to be accurate and robust. Importantly, these approaches are highly scalablewith low marginal cost, essentially matching human expert annotation in a near-automated workflow.Our results support the notion that transcriptomics combined with machine learning could be used aspart of an integrated -omics approach wherein risk prediction, differential diagnosis, and subclassifi-cation of AML are achieved by genomics while diagnosis could be assisted by transcriptomic-basedmachine learning.
For more information, see the original publication: Warnat-Herresthal, S. et al. Scalable Prediction of Acute MyeloidLeukemia Using High-DimensionalMachine Learning and Blood Transcriptomics. iScience 23, 100780, 2020. doi: 10.1016/j.isci.2019.100780
Differential Gene Expression in Circulating CD14+ Monocytes Indicates the Prognosis of Critically Ill Patients with Sepsis
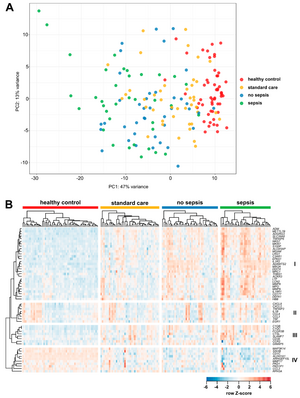
Critical illness and sepsis are characterized by drastic changes in the systemic innate immune response, particularly involving monocytes. The exact monocyte activation profile during sepsis, however, has remained obscure. Therefore, we prospectively analyzed the gene expression profile of circulating CD14+ monocytes from healthy volunteers (n = 54) and intensive care unit (ICU) patients (n = 76), of which n = 36 had sepsis. RNA sequencing of selected samples revealed that monocytes from septic ICU patients display a peculiar activation pattern, which resembles characteristic functional stages of monocyte-derived macrophages and is distinct from controls or non-sepsis ICU patients. Focusing on 55 highly variable genes selected for further investigation, arachidonate 5-lipoxygenase-activating protein (ALOX5AP) was highly upregulated in monocytes of ICU patients and only normalized during 7 days in the ICU in non-sepsis patients. Strikingly, low monocytic guanine nucleotide exchange factor 10-like protein (ARHGEF10L) mRNA expression was associated with the disease severity and mortality of ICU patients. Collectively, our comprehensive analysis of circulating monocytes in critically ill patients revealed a distinct activation pattern, particularly in ICU patients with sepsis. The association with disease severity, the longitudinal recovery or lack thereof during the ICU stay, and the association with prognosis indicate the clinical relevance of monocytic gene expression profiles during sepsis.
For more information, see the original publication: Liepelt A., Differential Gene Expression in Circulating CD14+ Monocytes Indicates the Prognosis of Critically Ill Patients with Sepsis. J. Clin. Med., 9(1), 127, 2020. doi: doi.org/10.3390/jcm9010127
Mind the Map: Technology Shapes the Myeloid Cell Space
The myeloid cell system shows very high plasticity, which is crucial to quickly adapt to changes during an immune response. From the beginning, this high plasticity has made cell type classification within the myeloid cell system difficult. This publication reports on earlier attempts of cell type classification in the myeloid cell system, discusses current approaches and their pros and cons, and proposes future strategies for cell type classification within the myeloid cell system that can be easily extended to other cell types.
For more information, see the original publication: Günther P. et al. Mind the Map: Technology Shapes the Myeloid Cell Space, Front Immunol, 10: 2287, 2019. doi:10.3389/fimmu.2019.02287
Transcriptional Signature Derived from Murine Tumor-Associated Macrophages Correlates with Poor Outcome in Breast Cancer Patients

Tumor-associated macrophages (TAMs) are frequently the most abundant immune cells in cancers and are associated with poor survival. Here, we generated TAM molecular signatures from K14cre;Cdh1flox/flox;Trp53flox/flox (KEP) and MMTV-NeuT (NeuT) transgenic mice that resemble human invasive lobular carcinoma (ILC) and HER2+ tumors, respectively. Determination of TAM-specific signatures requires comparison with healthy mammary tissue macrophages to avoid overestimation of gene expression differences. TAMs from the two models feature a distinct transcriptomic profile, suggesting that the cancer subtype dictates their phenotype. The KEP-derived signature reliably correlates with poor overall survival in ILC but not in triple-negative breast cancer patients, indicating that translation of murine TAM signatures to patients is cancer subtype dependent. Collectively, we show that a transgenic mouse tumor model can yield a TAM signature relevant for human breast cancer outcome prognosis and provide a generalizable strategy for determining and applying immune cell signatures provided the murine model reflects the human disease.
For more information, see the original publication: Tuit S. Transcriptional Signature Derived from Murine Tumor-Associated Macrophages Correlates with Poor Outcome in Breast Cancer Patients. Cell Rep. 29(5):1221-1235.e5, 2019. doi: 10.1016/j.celrep.2019.09.067.
Pheno-seq: Linking visual features and gene expression
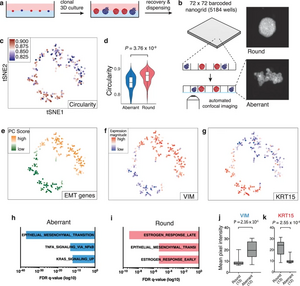
Linking heterogeneity of morphological phenotypes and the underlying transcriptome is still limited. “Pheno-seq” is able to directly link visual features of 3D cell culture systems with profiling their transcriptome. As prototypic applications breast and colorectal cancer (CRC) spheroids were analyzed by pheno-seq. We anticipate that the ability to integrate transcriptome analysis and morphological patho-phenotypes of cancer cells will provide novel insight on the molecular origins of intratumor heterogeneity.
For more information, see the original publication: Tirier, S.M.. et al. Pheno-seq – linking visual features and gene expression in 3D cell culture systems. Sci Rep 9, 12367, 2019. doi:10.1038/s41598-019-48771-4
Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases
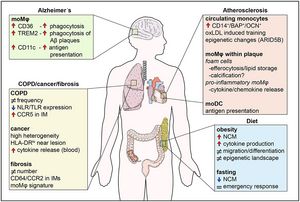
Human monocytes are divided in three major populations; classical (CD14+CD16−), non-classical (CD14dimCD16+), and intermediate (CD14+CD16+). Each of these subsets is distinguished from each other by the expression of distinct surface markers and by their functions in homeostasis and disease. In this review, we discuss the most up-to-date phenotypic classification of human monocytes that has been greatly aided by the application of novel single-cell transcriptomic and mass cytometry technologies. Furthermore, we shed light on the role of these plastic immune cells in already recognized and emerging human chronic diseases, such as obesity, atherosclerosis, chronic obstructive pulmonary disease, lung fibrosis, lung cancer, and Alzheimer's disease. Our aim is to provide an insight into the contribution of human monocytes to the progression of these diseases and highlight their candidacy as potential therapeutic cell targets.
For more information, see the original publication: Kapellos et al. Human Monocyte Subsets and Phenotypes in Major Chronic Inflammatory Diseases. Front. Immunol. 10: 2035, 2019. doi: 10.3389/fimmu.2019.02035
scGen predicts single-cell perturbation responses
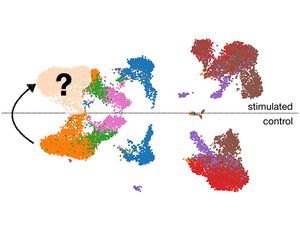
Scientists from the Helmholtz Zentrum München developed scGen that predicts single-cell pertubation processes.
Accurately modeling cellular response to perturbations is a central goal of computational biology. scGen (https://github.com/theislab/scgen) is a model, combining variational autoencoders and latent space vector arithmetics for high-dimensional single-cell gene expression data. scGen can accurately model perturbation and infection response of cells across cell types, studies and species. scGen learns cell-type and species-specific responses implying that it captures features that distinguish responding from non-responding genes and cells. With the upcoming availability of large-scale atlases of organs in a healthy state, we envision scGen to become a tool for experimental design through in silico screening of perturbation response in the context of disease and drug treatment.
For more information, see the press release and the original publication: Lotfollahi, M., et al. scGen predicts single-cell perturbation responses. Nat Methods 16, 715–721, 2019. doi:10.1038/s41592-019-0494-8
Current best practices in single‐cell RNA‐seq analysis: a tutorial
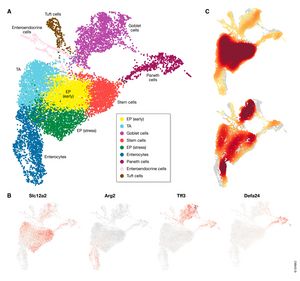
Single‐cell RNA‐seq has enabled gene expression to be studied at an unprecedented resolution. This review details the steps of a typical single‐cell RNA‐seq analysis, including pre‐processing (quality control, normalization, data correction, feature selection, and dimensionality reduction) and cell‐ and gene‐level downstream analysis. We formulate current best‐practice recommendations for these steps based on independent comparison studies. We have integrated these best‐practice recommendations into a workflow, which we apply to a public dataset to further illustrate how these steps work in practice. Our documented case study can be found at https://www.github.com/theislab/single-cell-tutorial.
For more information, see the original publication: Luecken M.D. et al. Current best practices in single‐cell RNA‐seq analysis: a tutorial, Mol Syst Biol 15, e8746, Mol Sys, 2019. doi:10.15252/msb.20188746
Single cell RNA-seq denoising using a deep count autoencoder
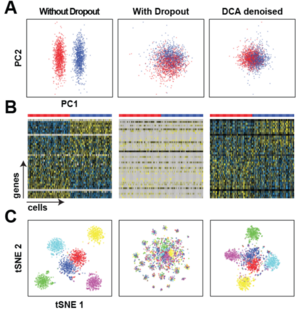
levels of dropout noise.
(c) Helmholtz Zentrum München
Scientists from the Helmholtz Zentrum München developed a deep count autoencoder (DCA) to denoise single cell RNA-seq datasets.
The deep count autoencoder network (DCA) denoises scRNA-seq data and removes the dropout effect by taking the count structure, overdispersed nature and sparsity of the data into account using a deep autoencoder with zero-inflated negative binomial (ZINB) loss function. DCA outperforms existing methods for data imputation in quality and speed, enhancing biological discovery. The software can be downloaded from: https://github.com/theislab/dca
For more information, see the original publication: Eraslan G. et al. Single cell RNA-seq denoising using a deep count autoencoder. Nat. Commun., 2019. doi:10.1038/s41467-018-07931-2
Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics
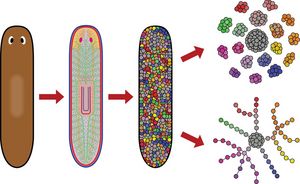
Flatworms of the species Schmidtea mediterranea are immortal—adult animals contain a large pool of pluripotent stem cells that continuously differentiate into all adult cell types. Therefore, single-cell transcriptome profiling of adult animals should reveal mature and progenitor cells. By combining perturbation experiments, gene expression analysis, a computational method that predicts future cell states from transcriptional changes, and a lineage reconstruction method, we placed all major cell types onto a single lineage tree that connects all cells to a single stem cell compartment. We characterized gene expression changes during differentiation and discovered cell types important for regeneration. Our results demonstrate the importance of single-cell transcriptome analysis for mapping and reconstructing fundamental processes of developmental and regenerative biology at high resolution.
For more information, see the original publication: Plass M. et al. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics, Science, 360(6391), 2018. doi:10.1126/science.aaq1723
Tracing the origin of each cell in a zebrafish
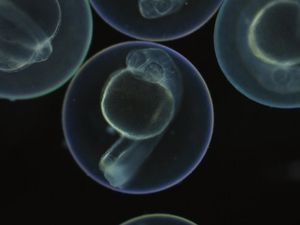
(c) Microscopic image: Junker Lab, MDC
Scientists from the Max Delbrück Center for Molecular Medicine in the Helmholtz Association (MDC) have used CRISPR-Cas9 genome editing to pioneer a technique capable of determining both the type and origin of all the cells in an organism.
"Whenever we use such a technology to examine an organ or an organism, we find not only familiar cell types, but also unknown and rare ones," says Dr. Jan Philipp Junker, head of the Quantitative Developmental Biology research group at MDC. "The next question is obvious: Where do these different types come from?" Junker's group describes a technique, called LINNAEUS that enables researchers to determine the cell type as well as the lineage of each cell.
For more information, see the press release by MDC, ScienceDaily, and the original publication: Spanjaard B. et al. Simultaneous lineage tracing and cell-type identification using CRISPR-Cas9-induced genetic scars. Nature Biotechnology, 2018. doi:10.1038/nbt.4124