Imputation of sparse data
Fabian Theis (HMGU), Maria Colomé-Tatché (HMGU), Alice McHardy (HZI) and Uwe Ohler (MDC) work on generating computational methods for the imputation of missing data from large scale data sets with specific noise models. For single cell transcriptomics expression levels of dropout genes (i.e., genes with missing expression data values) will be predicted using expression information of related cells. This is achieved within a deep learning framework using a deep autoencoder. For single cell genomics it has been shown that the direct sequencing of DNA of a single cell allows for calling copy number variations of genomic segments. Inference of single nucleotide (SNVs) requires whole genomic DNA pre-amplification, which introduces errors and biases and still large numbers of sites are devoid of information.
Develop and adapt missing-value invariant data mining methods
Emmanuel Müller (GFZ/HPI), Jörg Hackermüller (UFZ), and Alice McHardy (HZI) use a different strategy when dealing with sparse data compared to imputation of sparse data. They use downstream analysis methods that are invariant under missing values, denoted as missing value invariant data mining. Based on novel technologies in data mining they will be able to use and develop prediction, clustering and anomaly detection methods that are aware of the presence of missing values. Their methods will provide enhanced pattern detection compared to data imputation as data artefacts by pre-processing sparse data can be excluded. They developed similar methods in the past, for example missing value-invariant probabilistic nonlinear principal component analysis and fault tolerant subspace clustering for data with missing values. Here they propose to develop missing-value invariant methods for correlation network and lineage estimation.
Fusion with additional information (multi-view data)
Uwe Ohler (MDC), Maria Colomé-Tatché (HMGU), Fabian Theis (HMGU) and Alice McHardy (HZI) work on a project where single cell RNA-seq protocols have recently been complemented by assays that interrogate genomic and epigenomic data, either on the same cells or cells from the same population. These assays provide complementing, often (anti-)correlated information that should allow for a direct readout of the regulatory state of a cell. In practical terms, this means that we now have access to multiple distinct, but correlated data sets from the same (or from related) samples, offering different "views" of the cellular state. Each one of these datasets can be expected to only contain partial observations and considerable amount of missing data. Integrating these heterogeneous datasets and borrowing information across them will allow for better characterization of cellular states and possibly, better imputation of missing data from different individual omics layers.
Inclusion of spatial information
Roland Eils (DKFZ), Benedikt Brors (DKFZ), and Philipp Junker (MDC) work on spatial positioning of single cells, particluarly their relative location within a whole organ. This is a crucial parameter for understanding the functional context, the architecture of healthy tissues and organs as well as misregulation in aging and disease. Cellular heterogeneity and sparsity in tumor tissues for example can be responsible for therapy resistance and aggressive metastasis. Three-dimensional information can be obtained by advanced microscopy, but is typically unavailable in single-cell -omics. Here, the goal is to develop and extend methods to correlate spacial cellular localizations with single cell genomics information and hence impute the spatial contet. This will be used to add spatial information on top of single cell transcriptomics.
Co-design of computing infrastructure
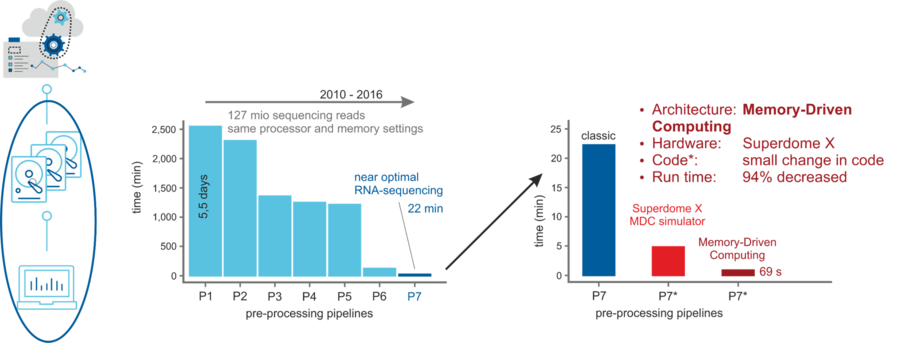
Dirk Pleiter (FZJ), Joachim Schultze (DZNE) together with industrial partners realize scalable architectures potimized for different parts of the workflow for single cell genomics. This will allow addressing problems with more than 105 parameters (genes) and more than 106 samples (cells), while still leaving potential for even larger problem sizes. The objective is to provide the design for future storage and computing infrastructure exploiting novel technologies, and setup of such an infrastructure initially for single cell imputation and later for other sparse2big users.
Generation of large test datasets and algorithm evaluation
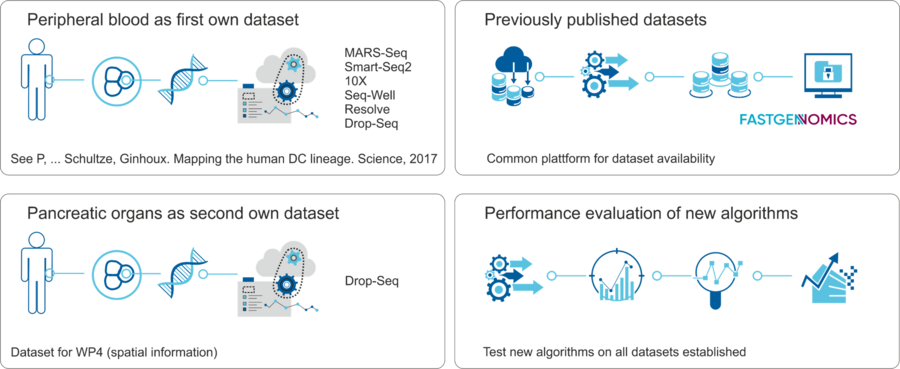
The development of new computational approaches will require access to single cell genomic datasets. We will use already existing datasets for evaluation and will provide unique novel data in combination with new computational approaches.